|
Paraparesi spastiche X-legate
549
riori; in tutti questi casi viene segnalato alla RM del
cranio un assottigliamento del corpo calloso.
SPG15/ZFYVE26
Il gene della SPG15 è stato originariamente mappa-
to in famiglie con la sindrome di Kjellin, una forma
complicata di paraplegia spastica con compromis-
sione cognitiva e maculopatia pigmentosa, ma dopo
l’identificazione del gene della SPG15, che codifica
per la proteina spastizina, il fenotipo correlato al de-
ficit genetico si è molto ampliato. Clinicamente la
SPG15 non può essere distinta dalla SPG11. Viene
descritto un fenotipo che può comprendere anche
neuropatia periferica, amiotrofia distale ed iperin-
tensità della sostanza bianca cerebrale alla RM del
cranio (Fig. 36.2A). Dal momento che è considera-
ta più rara dell’SPG11 (frequenza stimata < 3%), il
test genetico è raccomandato solo nei casi negativi
per SPG11 e se vi sono assottigliamento del corpo
calloso (Fig. 36.2B) e ritardo mentale. La mutazio-
ne SPG15 è stata trovata in circa 1/3 dei casi.
■
Paraparesi spastiche X-legate
SPG1/L1CAM
L’SPG1 è causata da mutazioni nel gene per la mo-
lecola L1 di adesione, L1CAM, responsabile di un
spettro fenotipico di malattia, la cosiddetta “sindro-
me L1”, comprendente molti disordini clinicamente
definiti. I segni clinici della sindrome L1 sono l’idro-
cefalo, identificato spesso già in epoca prenatale, se-
condario alla stenosi dell’acquedotto di Silvio, la di-
splasia, ipoplasia o aplasia del corpo calloso, il ritardo
mentale con ritardo nell’acquisizione del linguaggio,
la paraparesi spastica ed il pollice addotto. Il fenotipo
della SPG1 può variare anche all’interno della stessa
famiglia e non esiste una chiara relazione genotipo-fe-
notipo, sebbene le mutazioni tronche si leghino più
frequentemente ad un fenotipo grave con morte in età
precoce. Le donne portatrici sono raramente affette,
ma possono mostrare modesti segni clinici, quali pol-
lice addotto o lieve compromissione cognitiva.
Non vi sono dati che descrivano la frequenza della
mutazione L1CAM nel gruppo con fenotipo SPG1.
È stato visto, comunque, che la quantità di mutazio-
ni identificate in L1CAM aumenta con l’aumentare
del numero di segni caratteristici ed arriva all’85%
quando tre o più segni età-indipendenti (agenesia del
corpo calloso, idrocefalo, stenosi dell’acquedotto di
Silvio, pollice addotto) sono presenti. Il sintomo iso-
lato con più alto valore predittivo è la presenza del
pollice addotto (50%), seguita da paraparesi spastica
ed agenesia/disgenesia del corpo calloso (circa 40%).
SPG2/PLP1
L’SPG2 è causata da mutazioni nel gene
PLP1
,
codificante per la proteina proteolipidica 1. La
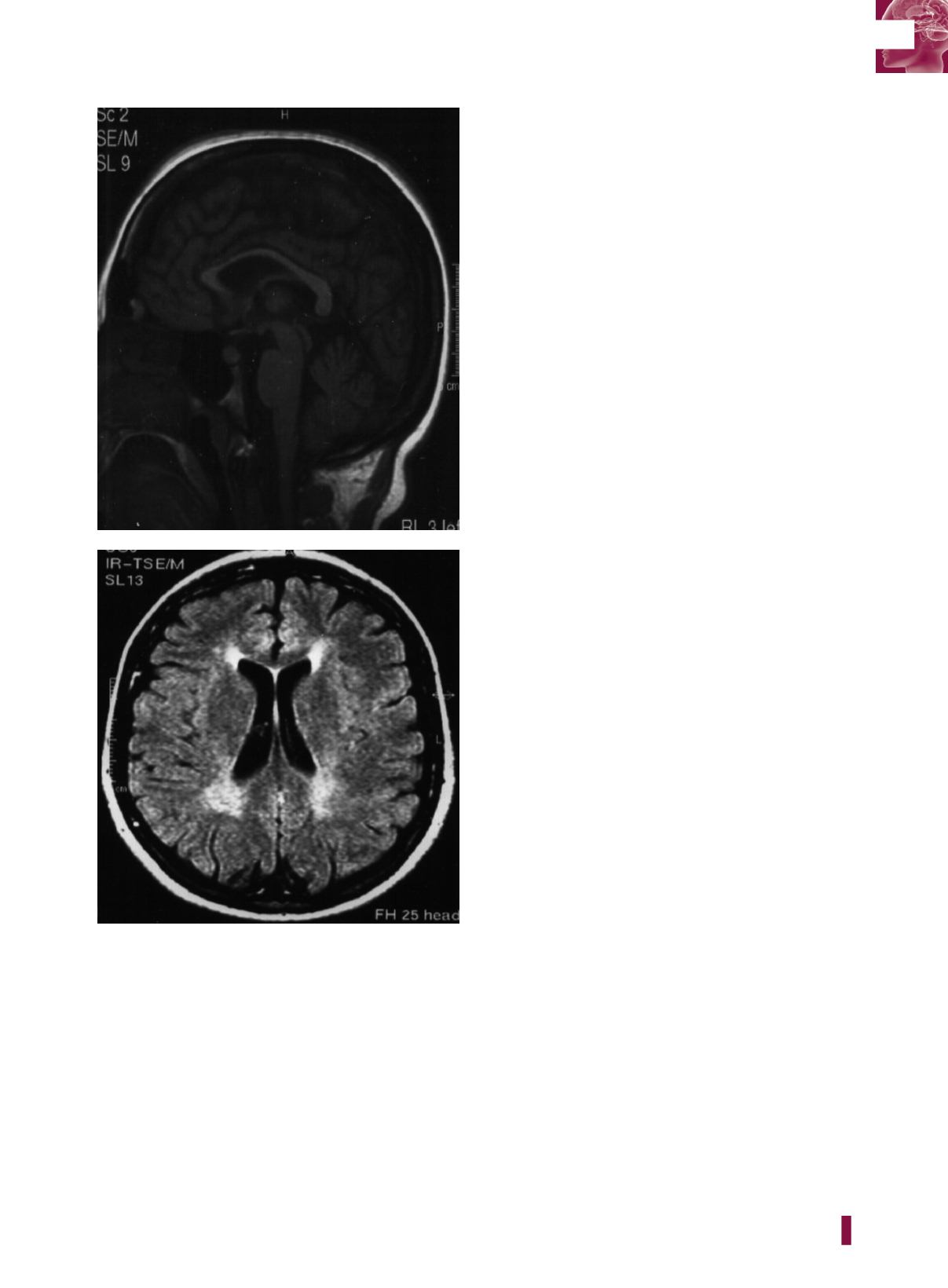
A
B
Figura 36
.
1
| (A) RM del cranio. Sezione sagittale, immagine
T1-pesata. Importante assottigliamento del corpo calloso. (B)
RM del cranio. Sezione assiale, immagine FLAIR. In sede pe-
riventricolare, in particolare intorno ai corni occipitali e frontali,
sono presenti zone di alterato segnale della sostanza bianca.


